| Reactivity | MuSpecies Glossary |
| Applications | ELISA |
| Conjugate | HRP |
| Background | The Quantikine Mouse PCSK9 Immunoassay is a 4.5 hour solid-phase ELISA designed to measure mouse PCSK9 in cell culture supernates, cell lysates, serum, and plasma. It contains NS0-expressed recombinant mouse PCSK9 and antibodies raised against the recombinant factor. This immunoassay has been shown to accurately quantitate the recombinant factor. Results obtained using natural mouse PCSK9 sh...owed linear curves that were parallel to the standard curves obtained using the Quantikine kit standards. These results indicate that this kit can be used to determine relative mass values for naturally occurring mouse PCSK9. Show More |
| Specificity | Natural and recombinant mouse PCSK9. This kit detects 60 kDa, 53 kDa, and LDLR-complexed recombinant mouse PCSK9. |
| Source | N/A |
| Assay Type | Solid Phase Sandwich ELISA |
| Inter-Assay | See PDF Datasheet for details |
| Intra-Assay | See PDF Datasheet for details |
| Spike Recovery | See PDF Datasheet for details |
| Sample Volume | See PDF Datasheet for details |
| Gene | Pcsk9 |
| Dilutions |
|
|
| Application Notes | No significant interference observed with available related molecules. |
|
| Publications |
|
| Storage | Store the unopened product at 2 - 8 °C. Do not use past expiration date. |
![Immunocytochemistry EGFR Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/EGF_R_AF231_Immunocytochemistry__Immunofluorescence_21143.jpg)
![Flow Cytometry EGFR Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/EGF_R_AF231_Flow_Cytometry_20401.jpg)
![Western Blot EGFR Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/EGF_R_AF231_Western_Blot_19925.jpg)
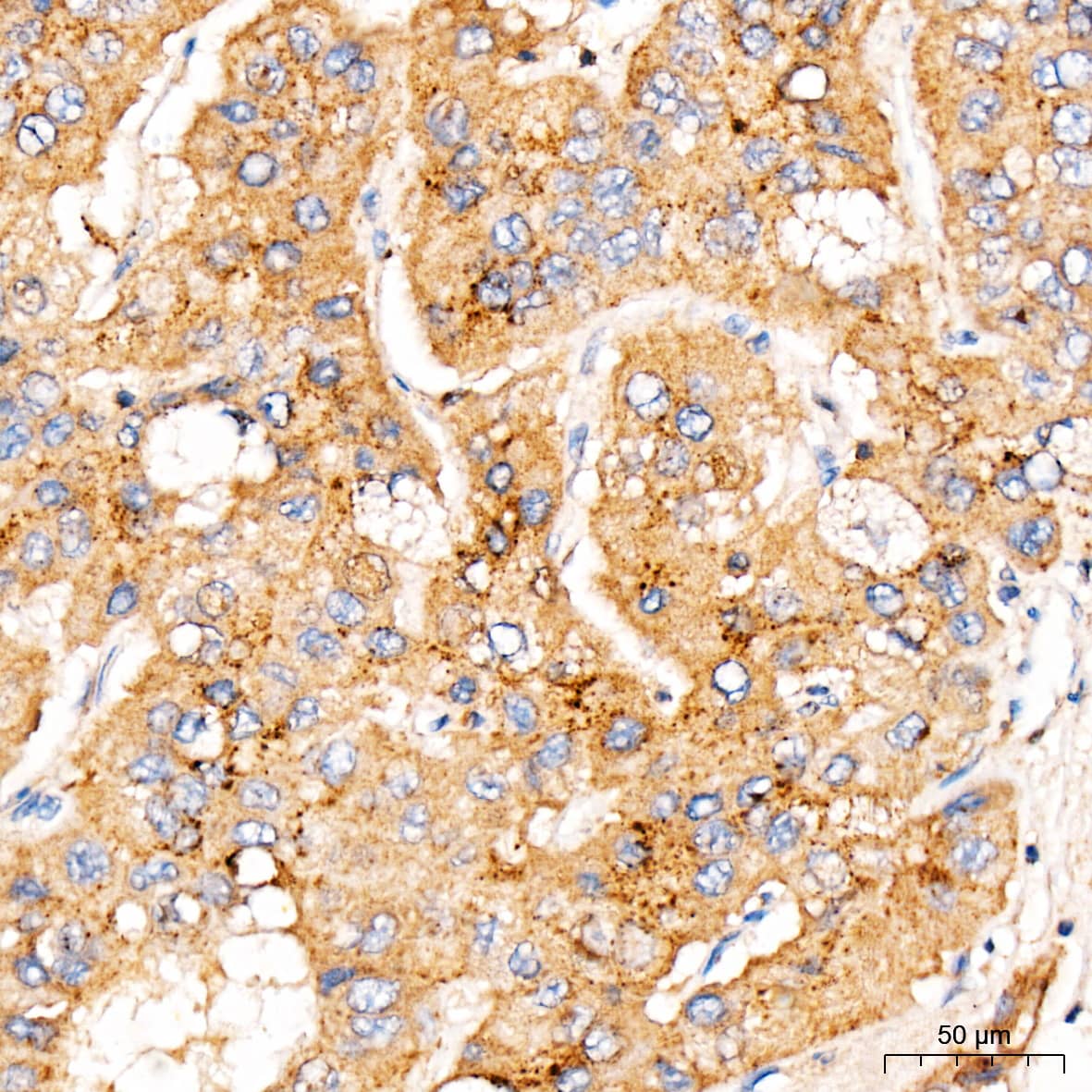
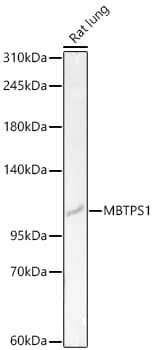
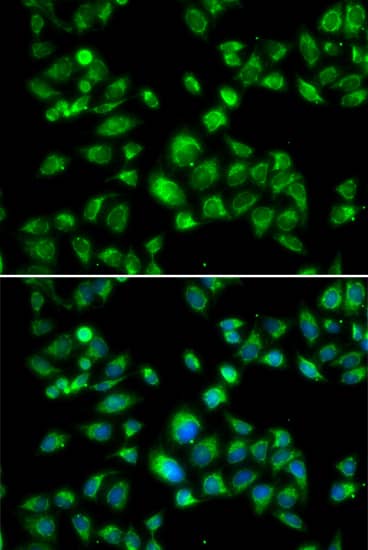

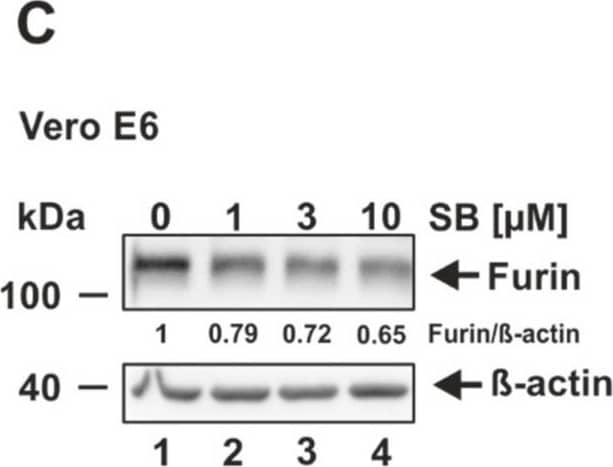





| Gene Symbol | Pcsk9 |




![Western Blot LDLR Antibody [Unconjugated]](https://images.novusbio.com/images/af2255_mouse-ldl-r-affinity-purified-polyclonal-ab-8120255541738.jpg)
![Western Blot LDLR Antibody [Unconjugated]](https://images.novusbio.com/images/af2255_mouse-ldl-r-affinity-purified-polyclonal-ab-8120255541731.jpg)
![Western Blot LDLR Antibody [Unconjugated]](https://images.novusbio.com/images/af2255_mouse-ldl-r-affinity-purified-polyclonal-ab-4120241240515.jpg)






![Immunohistochemistry Complement Factor H Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/Complement_Factor_H_AF4779_Immunohistochemistry_7182.jpg)
![Bioactivity EGF [Unconjugated]](https://images.novusbio.com/images/protein/EGF_236EG_1570.jpg)
![Cell Culture EGF [Unconjugated]](https://images.novusbio.com/images/236-eg_recombinant-human-egf-protein-cf-25202394526.jpg)
![Cell Culture EGF [Unconjugated]](https://images.novusbio.com/images/236-eg_recombinant-human-egf-protein-cf-bioactivity-25202394053.jpg)






![Immunohistochemistry Insulin Antibody (182410) [Unconjugated]](https://images.novusbio.com/images/antibody/mab1417_human-bovine-mouse-insulin-mab-clone-182410-immunohistochemistry-308202115145.jpg)
![Immunocytochemistry Insulin Antibody (182410) [Unconjugated]](https://images.novusbio.com/images/antibody/Insulin_MAB1417_Immunocytochemistry_9376.jpg)
![Immunocytochemistry CD55/DAF Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/CD55_AF2009_Immunocytochemistry__Immunofluorescence_23052.jpg)
![Immunohistochemistry CD55/DAF Antibody [Unconjugated]](https://images.novusbio.com/images/antibody/CD55_AF2009_Immunohistochemistry_6728.jpg)
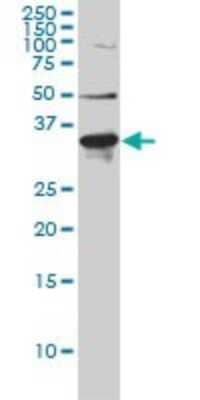
 followed by HRP-conjugated Anti-Goat IgG Secondary Antibody (Catalog # HAF019). A specific band was detected for Proprotein Convertase 9/PCSK9 at approximately 74 kDa (as indicated). This experiment was conducted under reducing conditions and using Immunoblot Buffer Group 7.")